Dehydrodihydrorotenone
PubChem CID
3950722
Structure
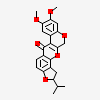

Molecular Formula
Synonyms
- Dehydrodihydrorotenone
- 16,17-dimethoxy-6-propan-2-yl-2,7,20-trioxapentacyclo[11.8.0.03,11.04,8.014,19]henicosa-1(13),3(11),4(8),9,14,16,18-heptaen-12-one
- KBio2_005802
- 16,17-dimethoxy-6-propan-2-yl-2,7,20-trioxapentacyclo(11.8.0.03,11.04,8.014,19)henicosa-1(13),3(11),4(8),9,14,16,18-heptaen-12-one
- Spectrum_000186
Molecular Weight
394.4 g/mol
Computed by PubChem 2.2 (PubChem release 2021.10.14)
Dates
- Create:2005-09-12
- Modify:2025-01-11
Description
LSM-4404 is a member of rotenones.
Chemical Structure Depiction
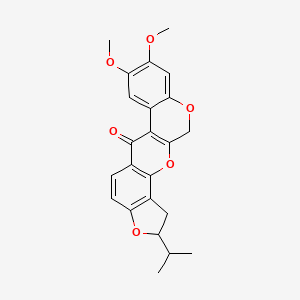
16,17-dimethoxy-6-propan-2-yl-2,7,20-trioxapentacyclo[11.8.0.03,11.04,8.014,19]henicosa-1(13),3(11),4(8),9,14,16,18-heptaen-12-one
Computed by Lexichem TK 2.7.0 (PubChem release 2021.10.14)
InChI=1S/C23H22O6/c1-11(2)16-8-14-15(28-16)6-5-12-22(24)21-13-7-18(25-3)19(26-4)9-17(13)27-10-20(21)29-23(12)14/h5-7,9,11,16H,8,10H2,1-4H3
Computed by InChI 1.0.6 (PubChem release 2021.10.14)
BHCBWJBSVOKHRJ-UHFFFAOYSA-N
Computed by InChI 1.0.6 (PubChem release 2021.10.14)
CC(C)C1CC2=C(O1)C=CC3=C2OC4=C(C3=O)C5=CC(=C(C=C5OC4)OC)OC
Computed by OEChem 2.3.0 (PubChem release 2024.12.12)
C23H22O6
Computed by PubChem 2.2 (PubChem release 2021.10.14)
- Dehydrodihydrorotenone
- 16,17-dimethoxy-6-propan-2-yl-2,7,20-trioxapentacyclo[11.8.0.03,11.04,8.014,19]henicosa-1(13),3(11),4(8),9,14,16,18-heptaen-12-one
- KBio2_005802
- 16,17-dimethoxy-6-propan-2-yl-2,7,20-trioxapentacyclo(11.8.0.03,11.04,8.014,19)henicosa-1(13),3(11),4(8),9,14,16,18-heptaen-12-one
- Spectrum_000186
- SpecPlus_000183
- Spectrum2_000184
- Spectrum3_000219
- Spectrum4_001437
- Spectrum5_000294
- BSPBio_001837
- KBioGR_001914
- KBioSS_000666
- DivK1c_006279
- SPBio_000107
- SCHEMBL4278197
- CHEMBL3039066
- CHEBI:93848
- KBio1_001223
- KBio2_000666
- KBio2_003234
- KBio3_001337
- LSM-4404
- CCG-38392
- LMPK12060066
- BRD-A33119430-001-02-2
- BRD-A33119430-001-03-0
- Q27165589
Property Name
Property Value
Reference
Property Name
Molecular Weight
Property Value
394.4 g/mol
Reference
Computed by PubChem 2.2 (PubChem release 2021.10.14)
Property Name
XLogP3-AA
Property Value
4.1
Reference
Computed by XLogP3 3.0 (PubChem release 2021.10.14)
Property Name
Hydrogen Bond Donor Count
Property Value
0
Reference
Computed by Cactvs 3.4.8.18 (PubChem release 2021.10.14)
Property Name
Hydrogen Bond Acceptor Count
Property Value
6
Reference
Computed by Cactvs 3.4.8.18 (PubChem release 2021.10.14)
Property Name
Rotatable Bond Count
Property Value
3
Reference
Computed by Cactvs 3.4.8.18 (PubChem release 2021.10.14)
Property Name
Exact Mass
Property Value
394.14163842 Da
Reference
Computed by PubChem 2.2 (PubChem release 2021.10.14)
Property Name
Monoisotopic Mass
Property Value
394.14163842 Da
Reference
Computed by PubChem 2.2 (PubChem release 2021.10.14)
Property Name
Topological Polar Surface Area
Property Value
63.2 Ų
Reference
Computed by Cactvs 3.4.8.18 (PubChem release 2021.10.14)
Property Name
Heavy Atom Count
Property Value
29
Reference
Computed by PubChem
Property Name
Formal Charge
Property Value
0
Reference
Computed by PubChem
Property Name
Complexity
Property Value
687
Reference
Computed by Cactvs 3.4.8.18 (PubChem release 2021.10.14)
Property Name
Isotope Atom Count
Property Value
0
Reference
Computed by PubChem
Property Name
Defined Atom Stereocenter Count
Property Value
0
Reference
Computed by PubChem
Property Name
Undefined Atom Stereocenter Count
Property Value
1
Reference
Computed by PubChem
Property Name
Defined Bond Stereocenter Count
Property Value
0
Reference
Computed by PubChem
Property Name
Undefined Bond Stereocenter Count
Property Value
0
Reference
Computed by PubChem
Property Name
Covalently-Bonded Unit Count
Property Value
1
Reference
Computed by PubChem
Property Name
Compound Is Canonicalized
Property Value
Yes
Reference
Computed by PubChem (release 2021.10.14)
177.94 Ų [M+H-H2O]+ [CCS Type: TW; Method: calibrated with polyalanine and drug standards]
185.29 Ų [M+H]+ [CCS Type: TW; Method: calibrated with polyalanine and drug standards]
Ross et al. JASMS 2022; 33; 1061-1072. DOI:10.1021/jasms.2c00111
194.6 Ų [M+H]+ [CCS Type: TW; Method: calibrated with polyalanine and drug standards]
Polyketides [PK] -> Flavonoids [PK12] -> Rotenoid flavonoids [PK1206]
Instrument Name
Jeol PS-100
Copyright
Copyright © 2002-2024 Wiley-VCH Verlag GmbH & Co. KGaA. All Rights Reserved.
Thumbnail
MoNA ID
MS Category
Experimental
MS Type
LC-MS
MS Level
MS2
Precursor Type
[M+H]+
Precursor m/z
395.1478
Instrument
Agilent 6530 Q-TOF
Instrument Type
LC-ESI-QTOF
Ionization Mode
positive
Collision Energy
40 V
Retention Time
5.975
Top 5 Peaks
279.0619 100
335.1288 76.20
293.0505 68.93
264.0438 61.28
280.0689 49.10
Thumbnail

MoNA ID
MS Category
Experimental
MS Type
LC-MS
MS Level
MS2
Precursor Type
[M+H]+
Precursor m/z
395.1478
Instrument
Agilent 6530 Q-TOF
Instrument Type
LC-ESI-QTOF
Ionization Mode
positive
Collision Energy
20 V
Retention Time
5.975
Top 5 Peaks
395.1425 100
364.1272 34.39
335.1271 30.55
363.1268 24.73
349.1043 8.68
Thumbnail

MoNA ID
MS Category
Experimental
MS Type
Other
MS Level
MS2
Precursor Type
[M+H]+
Precursor m/z
395.1489149
Ionization Mode
positive
Retention Time
5.917435442
Top 5 Peaks
363.124533 0.30
364.132466 0.25
335.129589 0.22
307.061484 0.06
339.08771 0.04
Thumbnail

MoNA ID
MS Category
Experimental
MS Type
Other
MS Level
MS2
Precursor Type
[M+H]+
Precursor m/z
395.1489149
Ionization Mode
positive
Retention Time
5.975075170054322
Top 5 Peaks
152.07097365532383 0.14
264.06548594517227 0.08
236.06422473917232 0.05
150.05612865532382 0.05
279.0912154101723 0.05
Thumbnail

Follow these links to do a live 2D search or do a live 3D search for this compound, sorted by annotation score. This section is deprecated (see here for details), but these live search links provide equivalent functionality to the table that was previously shown here.
Same Connectivity Count
Same Parent, Connectivity Count
Similar Compounds (2D)
Similar Conformers (3D)
Same Count
- CCSbaseCCSbase Classificationhttps://ccsbase.net/
- ChEBI
- ChEMBLLICENSEAccess to the web interface of ChEMBL is made under the EBI's Terms of Use (http://www.ebi.ac.uk/Information/termsofuse.html). The ChEMBL data is made available on a Creative Commons Attribution-Share Alike 3.0 Unported License (http://creativecommons.org/licenses/by-sa/3.0/).http://www.ebi.ac.uk/Information/termsofuse.htmlChEMBL Protein Target Treehttps://www.ebi.ac.uk/chembl/g/#browse/targets
- LIPID MAPSDehydrodihydrorotenonehttps://lipidmaps.org/databases/lmsd/LMPK12060066Lipid Classificationhttps://www.lipidmaps.org/
- MassBank of North America (MoNA)LICENSEThe content of the MoNA database is licensed under CC BY 4.0.https://mona.fiehnlab.ucdavis.edu/documentation/license
- Metabolomics WorkbenchDehydrodihydrorotenonehttps://www.metabolomicsworkbench.org/data/StructureData.php?RegNo=22712
- SpectraBase21,22-Dihydro-6,12a-didehydro-rotenonehttps://spectrabase.com/spectrum/LcAMn3vNimo
- Wikidata
- PubChem
- MolGenieMolGenie Organic Chemistry Ontologyhttps://github.com/MolGenie/ontology/
CONTENTS