|
|
|
PubMed | Entrez | Structure | PubChem | Help | |
| PubChem » PubChem Help » Sketcher Help | |||
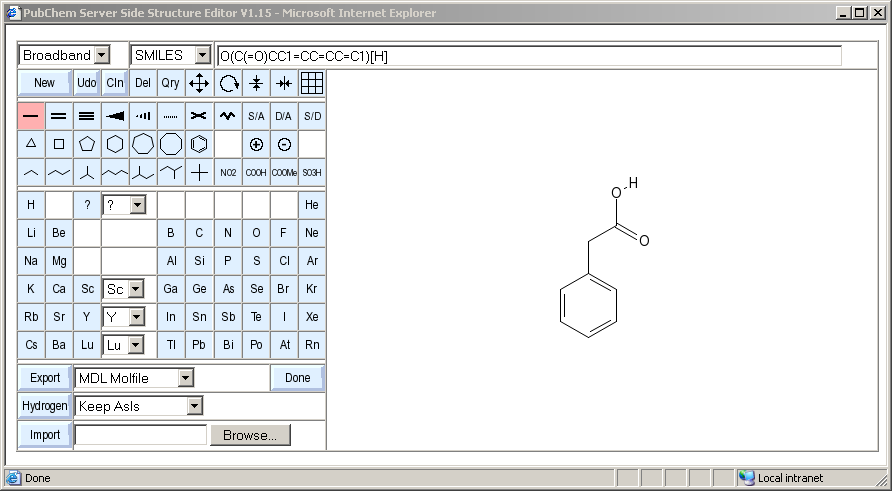
PubChem Sketcher Help
In order to allow input structures for queries, the PubChem Assay and Structure database uses its own unique Web-based sketcher tool. This document describes its features and operation.
System Requirements
|
Charges on existing atoms can be specified by selecting the plus or minus charge buttons. If one of these modes are active, a click on an existing atom will increase or decrease the charge by one.
Note that there is currently no support for specifying explicit radicals.
Below the bond drawing buttons, two rows of buttons allow the convenient input of larger structural fragments.
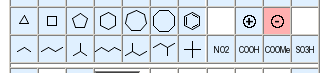
The first row of buttons display important basic ring systems. When a button has been activated, its associated drawing mode is used as follows:
A ring of the selected type is added, with its center at the click position.
The selected ring is sprouted from the atom via a single bond, using a 120 degree bond angle where possible.
The selected ring is sprouted from the atom, incorporating the start atom as first ring atom. If the start atom is already a ring atom, a spiro system is created.
The ring is annealed to the existing bonds. In case of the phenyl fragment, a smart decision is made about where to put double bonds in the added ring.
In case valence restrictions prevent the full execution of a ring addition, bonds to the source atoms may be omitted.
The second row of buttons displays a couple of important chain fragments and functional groups. These are used in a very similar fashion to the ring fragments, but the spiro or bond addition modes are not supported for them. They can only be added as stand-alone fragments or sprouted from an existing atom.
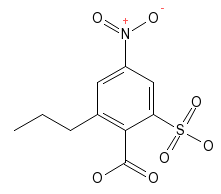
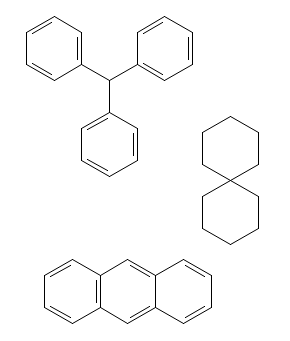
The structure to the right can be built with five mouse clicks into the drawing area, plus five button selections: select the phenyl ring fragment, click anywhere into empty drawing area, select the nitro functional group fragment, click onto the ring atom in the drawing to be substituted, select sulphonic acid group button, again click into drawing area, and repeat twice more for the carboxyl group and the n-propyl group. If desired, a complete set of hydrogen atoms can be added as a final step (see below).
The fragment button row on the main editor window only shows a small collection of frequently used fragments. A larger template library can be opened by clicking on the grid button in the upper right of the button section. An auxiliary window with tabs for various types of fragments of biological importance opens.
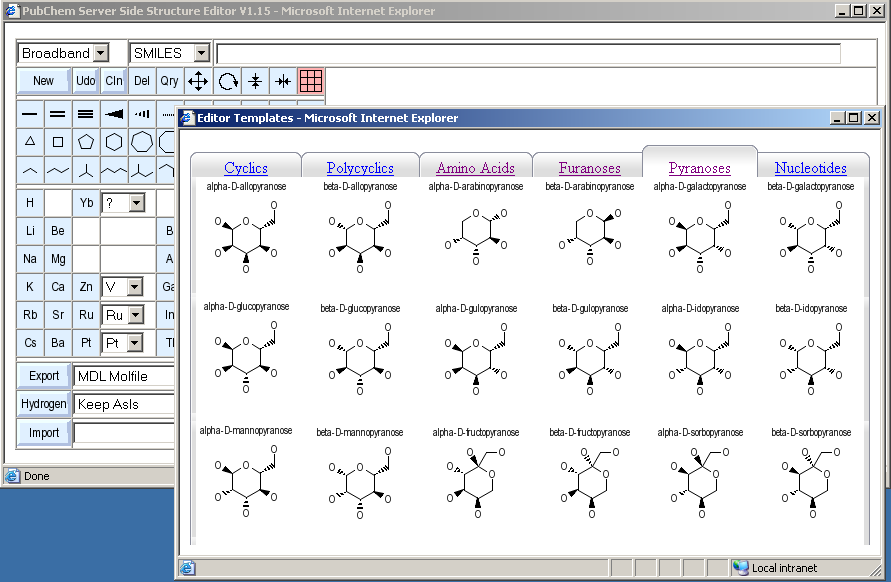
You can switch between template collections by clicking on the tabs. Individual templates are selected by first clicking onto them, and then into the drawing area where they should be placed. The click position is the center of the fragment placement position. After transferring a fragment into the drawing area, the template window is closed, and the sketcher automatically activates the move mode in order to allow more precise placement of the transferred fragment.
It is currently not possible to automatically link a transferred fragment to existing drawing components at the instant of transfer.
To the left of the template grid button, the top button row contains four buttons for graphical modifications of the current structure.
This mode allows you to move structure fragments, atoms or bonds. The position of the initial mouse click determines the object to be moved. If an atom is clicked, only that atom is moved around as long as the mouse key is pressed. All bonds to that atom will adjust. If the clicked object is a bond, both atoms of the bond will be moved in parallel. If neither an atom nor a bond is clicked, but the click point is within the bounding box of a larger fragment on the drawing area (a molecule), the whole fragment is moved.
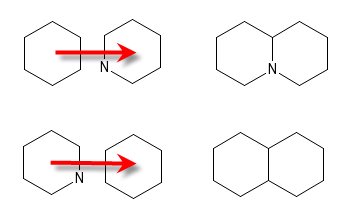
If at the moment the mouse button is released there are no overlaps between the moved atoms and any other atoms, only the graphical position of the moved objects will have been adjusted. If there are overlaps, an attempt is made to merge the overlapping atoms. Atoms that have not been moved have precedence. In the graphic to the left, if the left ring is moved onto the ring in the middle so that the rightmost two atoms of the moved ring overlap with the leftmost two atoms of the other ring, the results will be as depicted in the right column. If valence restrictions prevent some bonds from being formed, they will be omitted.
This mode will allow you to rotate fragments on the drawing area by clicking and dragging with the left mouse button. The center of rotation depends on the object at the location where the mouse button was clicked. It can be either an atom, a bond (center of rotation is the center of the bond) or a molecule when the click occurred in the bounding box (the center of rotation is the molecule center). Rotation is currently locked to 30 degree steps. When the rotation is finished, after releasing the mouse button, an atom merging step identical to that in the move mode is performed.

The mirror buttons allow you to easily generate the mirror image of a fragment, or to change the stereochemistry of a double bond. If a double bond is clicked, the ligands on one side of the bond are flipped so that the compound with opposite cis/trans stereochemistry on that bond results. If the click point is a molecule box, the whole molecule is mirrored along the x or y axis. All tetrahedral stereochemistry and wedge bonds are updated to represent the enantiomer of the mirrored molecule. There is no special action for clicking onto an atom - this is the same as a click into the bounding box.
The button marked Del is used to enter the object deletion mode. When this mode is active, the following operations are supported:
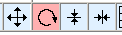
The atom and all the bonds it participates in are deleted.
The bond will be deleted, but its atoms remain.
The complete fragment will be deleted.
This is shown in the image above. A red box is displayed which follows the dragged mouse. When the mouse button is released, all objects within the selected area are removed.
If you are using a mouse with more than one button, the right mouse button is a shortcut to deletion opeations. It will always work, without the need to switch into the deletion mode. It supports the quick deletion of atoms, bonds and full fragments. The selection rectangle can only be used in the proper deletion mode.
The quick deletion mode is especially useful when you needed to click into the drawing area, for example in order to assign it the keyboard focus, and by this click inadvertently added a single atom. A quick right click, and the spurious addition is gone.
The button marked Udo implements a simple undo/redo facility. Only a single operation can be undone. If the button is then clicked again, the undo operation is itself undone, i.e. you end up with the old structure again.
The New button deletes the current drawing completely and gives you a blank slate. This operation can also be undone in case the command was executed in error.
Both buttons perform their operations immediately (as indicated by their raised shape) and are not modes. Undo does not change the current sketcher mode, New resets it to the single bond drawing tool.
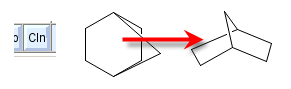
The button labelled Cln (clean) recomputes the structure layout without changing other aspects of the structure. The image to the right shows a sample molecule before and after cleanup. In case a cleanup should not yield an improved structure layout, it can be undone.
The sketcher supports the setting and deletion of a limited set of query attributes on atoms and bonds. In order to activate the query attribute mode, click the Qry button. In this mode, the left mouse button can be used to click onto atoms or bonds. Depending on the type of object clicked, different query attribute windows will open.
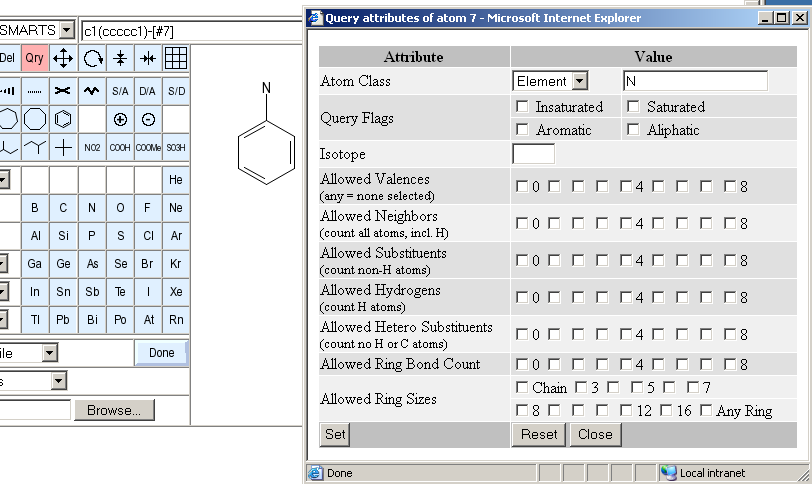
The image above shows the atom attribute window. The following attributes can be set or reset:
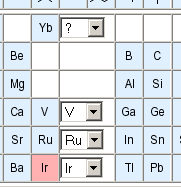
There are four different classes and three predefined element sets accessible in this row. By default, atoms in the sketch correspond to a specific type of physical atom with a defined element. If this atom class is selected, the element may be changed by entering a new element symbol in the text filed to the right. This is equivalent to selecting an element button and clicking onto the atom which should be changed. The other atom classes are any, list and negative list. An any atom will match any atom in database structures when used in substructure queries. The text field is ignored for this atom type. A list is a set of alternative elements for this atom. You need to specify a whitespace or comma-separated list of acceptable element symbols in the input field to the right. The negative list is the same as a normal list, except that all elements except the ones specified in the text field will match. Finally, the predefined element sets hetero, halogen and metal are shortcuts for popular element lists. These shortcuts also ignore the text field. Atom classes other than the simple element atom cannot be used for hashcode-based full structure searches.
This set of four check boxes allows you to request saturation or insaturation, and explicit aliphatic or aromatic character of a matched database structure atom. Saturated/insaturated and aliphatic/aromatic are mutually exclusive. These explicit flags which are applied only to a single atom always override any global match conventions set in a general structure search panel which opened the editor window.
Here you can specify the nucleon count of an isotope label on that atom. The input is a single integer. This information will automatically be used for substructure searches and also, if an appropriate hashcode is used, for hashcode-based full-structure queries.
This is a set of checkboxes where you can set allowed valence states of the atom when matched to a database structure. If no explicit valences are selected, the atom can be of any valence. Note that non-VB bonds (complex bonds, ionic bonds, etc.) in database structures have a zero valence count contribution.
This checkbox set works the same way as the valence row above, except that the bonded neighbors are simply counted, and bond orders and bond types are ignored.
Essentially the same as the neighbor count, except that hydrogen is not counted in the bonded neighbors.
This constraint is complementary to the substituent and neighbor counts.
Essentially the same as the neighbor count, except that neither hydrogen nor carbon bonded neighbors are counted.
The number of ring bonds the atom may participate in. Note that both VB and complex bonds can be ring bonds, but ionic bonds and other exotic types are excluded in the ring bond detection and are never considered part of a ring.
If you select one or more of these checkboxes, the atom can only match to database structure atoms which are a member of a ring of at least one of the selected sizes, or specifically a chain atom. The ring set used to determine this property is dependent on the back-end structure database system. For PubChem, it is an extended set of smallest rings in which all 3-atom sequences of bonded ring atoms are part of at least one ring. This ring set is more symmetrical than the classical SSSR.
Any atom query attributes which are set in the atom query panel, or which are already present when a structure is pre-loaded or imported, are reflected in the drawing on the canvas.
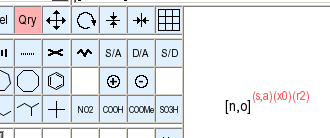
Atoms with query attributes are drawn with extended atom symbols and/or read attribute annotations. These special element symbols are used for extended symbols:
For query attributes with alternative possible counts, an attempt will be make to contract the displayed set as much as possible using closed ("1-2") and open ("-1" or "4-") ranges and lists of alternative single values ("3,5").
The structure handling library used to implement this applications supports many more query attributes than those accessible via the atom attribute panel. In case these were imported by reading a file with a query specification, other attributes may be displayed in addition to those described in this section. As long as they are not overwritten by explicit setting of new attributes on affected atoms, they will, if possible in the structure data transfer format, be preserved in structures submitted via the editor.
If the editor is in query attribute mode, and you click onto a bond, this query attribute window opens:
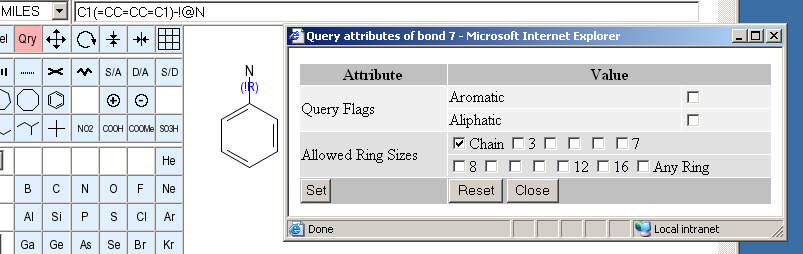
Its mode of operation is very similar to the corresponding atom panel, but there are different and fewer attributes which can be set and changed.
You can request a bond to be matched only to aromatic or aliphatic database structure bonds.
If you select one or more of these checkboxes, the bond can only match to database structure bonds which are a part of a ring of at least one of the selected sizes, or specifically a chain bond. The ring set used to determine this property is dependent on the back-end structure database system. For PubChem, it is an extended set of smallest rings in which all 3-atom sequences of bonded ring atoms are part of at least one ring. This ring set is more symmetrical than the classical SSSR.
As for atoms, query attributes of bonds are displayed on the drawing area. The annotation color for bond attributes is dark blue, and attribute annotations are drawn over the center of the bond.
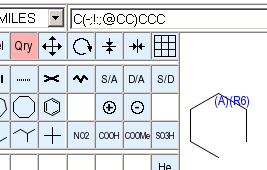
These characters are used to display bond query attributes:
For query attributes with alternative possible counts, an attempt will be make to contract the displayed set as much as possible using closed ("1-2") and open ("-1" or "4-") ranges and lists of alternative single values ("3,5").
The structure handling library used to implement this applications supports many more query attributes than those accessible via the bond attribute panel. In case these were imported by reading a file with a query specification, other attributes may be displayed in addition to those described in this section. As long as they are not overwritten by explicit setting of new attributes on affected bonds, they will, if possible in the structure data transfer format, be preserved in structures submitted via the editor.
Above the drawing area, a text field displays continuously updated information about the currently edited structure. The type of data displayed can be changed by the choice menu to the left of the text field.
The following choices are available:
The text line shows a SMILES encoding of the edited structure, assuming that hydrogens are implicitly added. The encoding of aromatic systems is in Kekul� form for maximum easy of decoding.
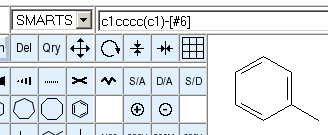
The text line displays a SMARTS encoding. The atom aromaticity atom attribute (lowercase element symbols) is automatically set for identified aromatic systems in the drawing. Atoms for which the aromaticity status cannot be determined are encoded as element numbers ([#6] for carbon) to avoid implicit assumptions about their aromaticity when decoding. Aromatic bonds are encoded as implicit bonds, aliphatic single bonds use explicit single bond encoding ([#6]-[#6]).
The line displays an InChI encoding of the structure. InChI is a new IUPAC standard for compact, unique representation of chemical structures. A full set of implicit hydrogen is assumed in the encoding. All hydrogen atoms are encoded with fixed positions so that the structure decodes to exactly the same tautomer as drawn.
The line displays the molecular formula and molecular weight. A full complement of hydrogen atoms is implicitly assumed.
The line content is a Sybyl Line Notation encoding of the current structure.
The text in the data field can be conveniently be copied and pasted with normal text highlight and clipboard operations. Thus, the sketcher can be a convenient input tool even for sites which do not possess a direct data update link to the main query form but do allow their structure input data to be encoded in SMILES, SMARTS or SLN.
In the SMILES, SMARTS and SLN representations, atom and bond query attributes are encoded as far as technically possible. Not all supported query attributes can be expressed in all of these line notation formats. The sketcher SMILES encoding uses the following custom extensions:
The structure data line serves not only as an information display. It is possible to enter structure codes in this field and import structure data into the drawing area.
Structure import is performed by clearing the line, and then editing or pasting a structure code into this field and finally pressing the return key. The setting of the display choice menu to the left of the field has no influence on the operation.
If the decoding of the structure data succeeds, the encoded content will be merged into the existing drawing as a new additional fragment. If it is intended to replace the current contents, you need to clear the drawing area first by pressing the New button. Fragments are imported without implicit hydrogens and are placed automatically.
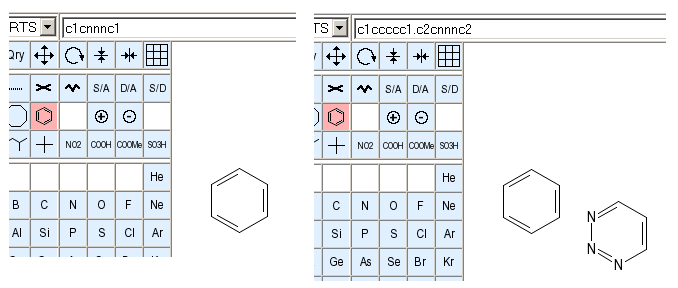
The images above display the drawing area before and after the SMILES string c1cnnnc1 was imported. Note that the implicit aromatic system in the input string was automatically resolved to a proper Kekul� form.
The complete set of supported structure data string formats:
The supported strings that can be imported via the data line may also be directly pasted from the clipboard into the drawing area by means of the standard ctrl-V keyboard shortcut, provided that the drawing area has keyboard focus. This method has the additional advantage that the location of the mouse at the moment of the keypress determines the location of the center of the newly added fragment. Depending on the browser and client operating system, the drawing area may not automatically have keyboard focus when you move the mouse into it. To make sure that it has, click the mouse once. In case this leads to the addition of an unwanted atom or a fragment, use the right mouse button to delete these.
The final method for loading existing structure data into the sketcher is by means of file upload.
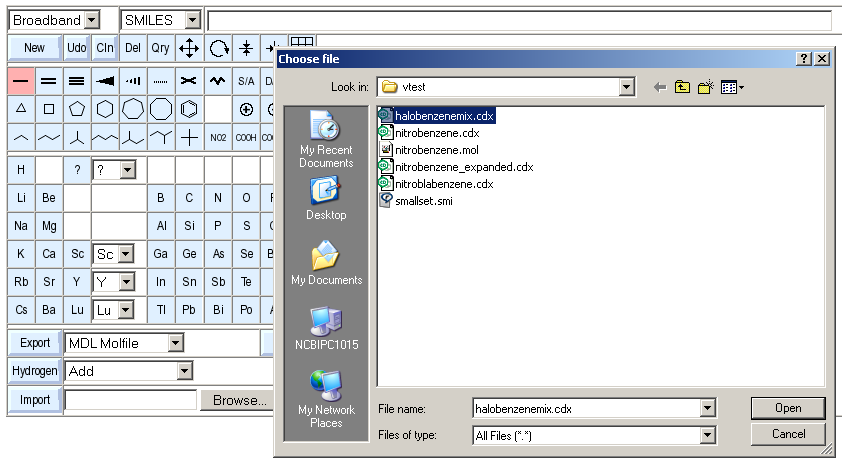
In order to do this, select an existing structure file via the Browse.. button and then press the Import button. The file is then read and added to the existing content. In case you want to guarantee that the imported file is the only sketcher content, press the New button before the import.
The import function only reads the first record of multi-record files. So in case you attempt to upload an SD-file, only the first record will show.
The hydrogen status of the imported structure will be adjusted at upload time depending on the setting of the Hydrogen option menu. These are its possible values:
A standard set of hydrogens is added to all open valences.
Hydrogen is added to all hetero atoms and carbon atoms where it is needed to make the encoding unambiguous, i.e. at stereo centers and stereo bonds, as well as to carbon atoms which traditionally are drawn with explicit hydrogens (aldehydes, C triple bond terminals, etc.).
The hydrogen status is kept as it was in the upload file.
Hydrogen is remove from carbon atoms, except where it is needed to determine stereochemistry, or where it is traditionally drawn (aldehydes, C triple bond terminals, etc.)
All hydrogen atoms are removed from input.
The sketcher can export the currently edited structure in various formats. This capability makes it a convenient tool for the input of data sets of limited to be used locally, or even file format conversion.
The current structure is exported by clicking on the button labelled Export. A file selector box will open and let you specify the name and location of the file downloaded from the sketcher server.
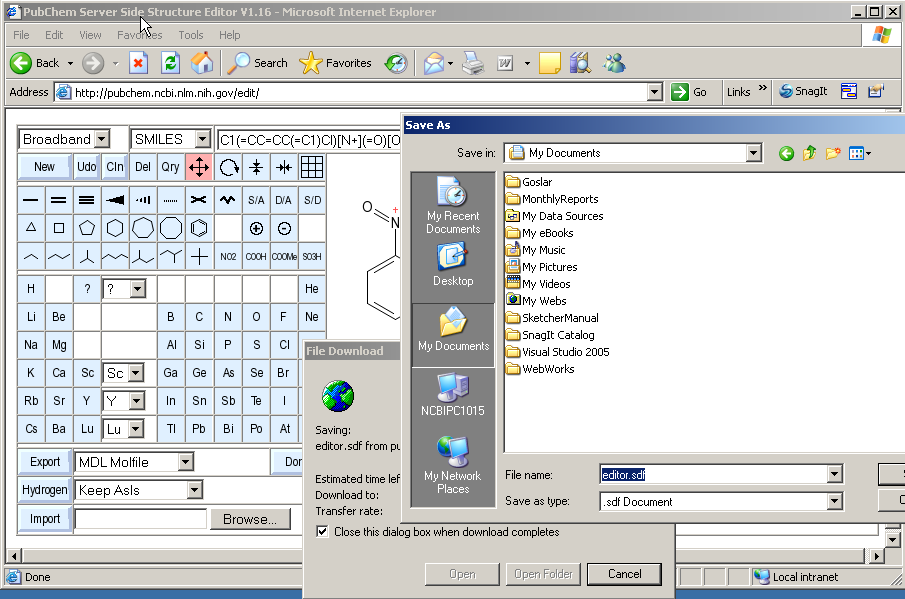
The default name of the download file is editor.xxx, where xxx is a suitable default suffix for the selected file format.
The desired format of the file can be selected, before clicking the Export button, from the menu to the right of that button. The exact list of formats which can be used for export is : structure exchange formats (MDL Molfile, SDF, SMILES, SMARTS), structure editor formats for further clean-up of the drawings for publications etc. (ChemDraw CDX) and image downloads (GIF, PNG, SVG, EPS).
Besides its role for file import and export of structure data, the Hydrogen option menu can also be used for direct manipulation of the currently edited structure.
Simply set the menu to the desired operation, and press the Hydrogen button to the left of the menu. The hydrogen status of the drawn structure will be immediately adjusted.
Many of the sketcher modes can be controlled by keyboard shortcuts. This feature allows advanced users to keep the mouse in the drawing area without moving it left and right to switch buttons.
These are the shortcuts:
The mnemonic for element shortcuts is that lowercase letters select the element where the single-letter symbol corresponds to the pressed key. Uppercase letters select the most important element with a two-letter symbol which starts with the pressed key.
There is no 9-membered ring template which could be associated with key 9.
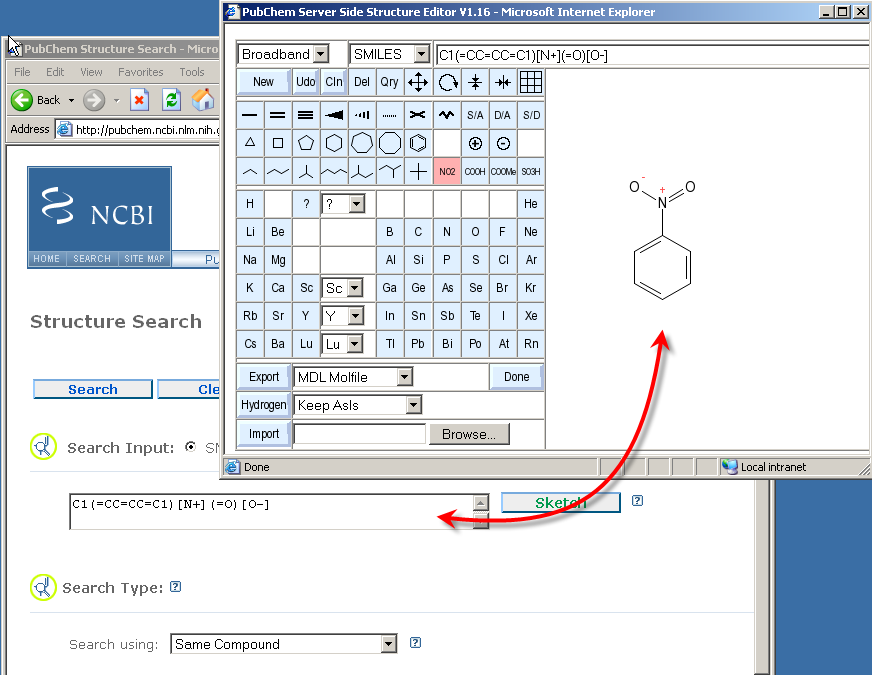
A question which has been asked more than once concerns the problem of how to transfer the edited structure data from the sketcher to a linked form which opened the sketcher window.
The answer is simple: There is absolutely nothing a user needs to do to achieve this. Data transfer is automatic, and dynamic. Every structure change is immediately reported to the caller form. There is no button which needs to be clicked in order to transmit the currentl sketch.
As described above, there is no need for any user action to transmit the structure data to an originating form for further processing. The sketcher can thus be quit at any time, without fear of data loss, simply by closing its window by means of the standard mechanisms of the client platform, such as clicking on the cross-shaped close icon on the upper right of the windows on MS Windows.
Nevertheless, since in our experience many users appear to be more comfortable if they can hit a dedicated, clearly labelled button to finish the arduous task of inputting an important query structure, we have added a big Done button to the sketcher button set. It is located to the right of the Export controls.
This prominent button simply closes the sketcher window and does nothing else.
| Write to Helpdesk | Disclaimer | Privacy statement | Accessibility | |