PubChem 3D Viewer v2.0
|
Overview |

|
PubChem 3D Viewer provides a user friendly interface for rendering multiple 3-dimensional structures of
PubChem compound records and for visualization of structure conformer overlays. Highly customizable 3D rendering
engine of this application provides dynamic molecular visualization experience together with
ability to create high resolution, publication quality images.
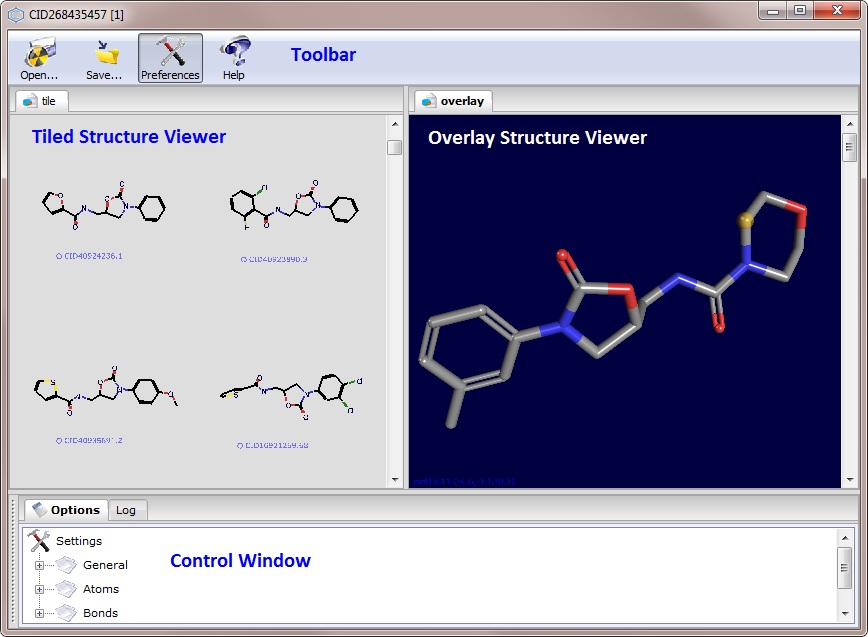
| Application Interface |
|
PubChem 3D Viewer consists of several interface elements providing different views of 3D structures and
organizing acces to multiple commands and options in the program. The application window
is split into three sections: the tabbed 3D Viewers on the left, the tabbed Controls section on the right
and the Information section at the bottom of the application window.
 |
- Toolbar
- Open...
- Open a new molecule file - loads a new molecule from one of the supported file formats. Note that the compounds loaded
before this operation will be discarded.
- Add molecules into your environment - loads more molecules from files, but maintains the previously loaded conformers in the PC3D Viewer.
- Close molecule files - closes opened molecule files.
- Save...
- Save current molecules into a molecule file - saves current molecules into a PubChem 3D file.
- Save current overlay view as an image - saves current overlay view into an image file.
- Preferences
- shows/hides Control Windows.
- Help
- displays this page.
- Overlay Structure Viewer
- allows users to see single molecular structure or to
overlay multiple structures in a single coordinate system. This view is
ideal for observing a single molecule in detail and/or for looking at
structural overlap of different conformers to see similarities and differences
in molecular structures.
- Tiled Structure Viewer
- provides ability to show multiple molecules in
different tiled sections of the viewer. This view is designed to browse
multiple conformers and analyse overall 3D coverage of conformer space.
Double clicking on any given conformer tile will show that particular conformer in
an overlap view.
Note, that this viewer is enabled only if multiple conformers are loaded into the PC3D Viewer.
- Control Windows
- Options
- provides ability to customize the appearance of
the graphical elements in molecular visualization and tune the behavior of the PC3D Viewer.
Clicking (+) or (-) signs on the tree node allows access to preferences arranged hierarchicaly into
three main groups:
- General - controls rendering options affecting the appearance of the molecule as a whole.
- Image Quality - higher values generate images of greater qualities, but may decrease
the speed of keyboard and mouse rotations.
- Light Model - allows to use different lighting on the molecular scene. One may toggle the
ambient light, emmisivitiy of atomic and bond surfaces, add or remove highlights and shininess.
- Background Color - specifies the color of the 3D viewer window not occupied by the molecule.
- Viewing Angle - controls the amount of perspecive in the projection. Higher values are equivalent
to looking from the objective of an ultra-wide angle camera and show nearby objects scaled bigger and
further objects appearing smaller. Smaller values of viewing angle turn the appearance of the molecule into
flatter projection with less perspective scaling.
- Scale Strategy - toggles between different scaling modes of the molecule into the viewing area. User may chose not to scale, or scale only once at the loading time of the molecule, or to scale relative to the first conformer shown, or use all conformers for scaling.
- Orthogonal Projection - toggles between perspective and orthogonal modes of projection. In orthogonal
mode objects appear at the same scale regardless of distance; in perspective mode further objects are scaled down.
Shortcut 'O'-key.
- Hide Coordinate System - toggles the visibility of reference coordinate system. Shortcut 'C'-key.
- Hide Box - controls the visibility of virtual box containing the molecule. Shortcut 'X'-key.
- Transparent Conformers - toggles the transparency of conformer overlaps. Note, that the reference
conformer (the first visible) is always shown opaque regardless of this flag. However, the following conformers are
affected by this flag.
- Show Pharmacophores - toggles the visibility of pharmacophores. Shortcut 'G'-key.
- Show Mol-Info - toggles the visibility of molecular information in tiled structure viewer. Shortcut 'I'-key.
- Conformer Opacity - defines how transparent are the non-reference conformers. Lower values of opacity correspond to transluscent
appearance and higher values make query conformers look more solid.
- Pharma Scale - determines the relative size of Pharmacophores.
| 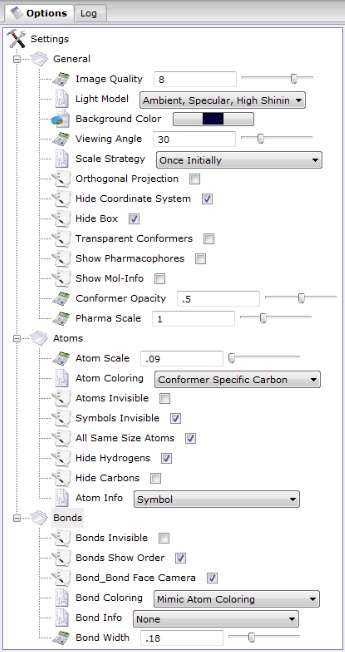 |
- Atoms - includes options affecting the appearance of chemical elements in the drawing.
- Atom Scale - determines the relative size (radius) of atomic spheres.
- Atom Coloring - controls the color schema for rendering atoms:
- Element Specific - uses conventionally accepted coloring for elements: Carbons as Gray, Oxygens as Red, Nitrogens as Blue, etc...
- Conformer Specific - colors all atoms of molecules to the same color, specific to the particular conformer. Conformer colors are assigned
at the loading time. These are predefined enumerated colors designed to best represent differences in conformer overlaps.
- Conformer Specific Carbon - carbons follow conformer specific coloring mode, all other atoms follow element specific schema.
- Black Carbon - renders carbons as black spheres, other atoms are colored according to their element specific color.
- Non Specific - colors all atoms in a single light gray.
- Atoms Invisible - toggles the visibility of atomic spheres. Shortcut 'A'-key.
- Symbols Inivisible - controls the visibility of atomic Symbols. Shortcut 'S'-key.
- All Same Size Atoms - dictates to render atomic spheres uniformly, not proportional to their covalent radii.
- Hide Hydrogen - toggles the visibility of Hydrogen atoms. Shortcut 'H'-key.
- Hide Carbons - toggles the visibility of carbon atoms. Shortcut 'Q'-key.
- Atom Info - toggles the information shown on atomic spheres. User may choose to show corresponding
chemical element's symbol, periodic number of the element, the serial number of atoms in the conformer, the
charge or the isotopic mass of the element.
- Bonds - consists of preferences influencing the appearance of bonds.
- Bonds Invisible - controls the visibility of bonds' cylinders. Shortcut 'B'-key.
- Bonds Show Order - toggles the appearance of multiple bonds as single cylinder or a pack
of thinner cylinders, emphasizing the higher bond orders.
- Bond_Bond Face Camera -
- Bond Coloring - controls the coloring of bonds.
- Element Specific - folows conventionally accepted coloring for elements: Carbons as Gray, Oxygen as Red, Nitrogen as Blue, etc...
- Conformer Specific - colors all atoms of molecules to the same color specific to the particular conformer. Conformer colors are assigned
at the loading time. Conformer colors are predefined enumerated colors designed to best represent differences in conformer overlaps.
- Conformer Specific Carbon - carbons follow conformer specific coloring mode, all other atoms follow element specific schema.
- Black Carbon - renders carbons as black spheres, other atoms are colored according to their element specific color.
- Non Specific - colors all atoms in a single light gray.
- Mimic Atom Coloring - makes the coloring of bonds similar to coloring of atoms, whatever the choice.
- Bond Info - selects the information shown on bonds. User may choose to see bond lengths or bond orders.
- Bond Width - determines the relative thickness of bonds cylinders.
- Log
- is designed to provide information about
the molecular structure, and about success or failure of operations performed. The content of
this window can be copy/pasted, edited and manipulated manually to create a operation journal.
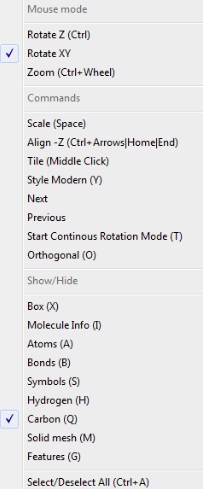
- Context Menu
- organizes easy access to the most
frequently used commands, provides shortcuts and brief description of commands' behavior. Commands
are arranged into three main groups:
- Mouse Move
- Rotate Z - rotates around Z axis.
- Rotate X/Y - rotates around X and Y axes.
- Zoom - zooms in or out.
- Scroll - scrolls up or down in tile viewer.
- Commands
- Scale - toggles between different scaling modes of the molecule into the viewing area.
User may chose not to scale, or scale only once at the loading time of the molecule,
or to scale relative to the first conformer shown, or use all conformers for scaling.
- Align - operations aligning the molecule along predefined axes.
User may chose to align the compounds along positive (+) or negative (-) direction
of different principal axes of intertial coordinate system.
- Tile - commands toggling between overlay and tile viewer.
- Style - commands toggling the predefined rendering styles.
- Next - selects the next molecule in tile viewer.
- Previous - selects the previous molecule in tile viewer.
- Continous Rotation Mode - toggles continous rotations mode. In this mode the very last mouse rotation operation is repeated continoutly until the user stops either by pressing Escape or by using the shortcut 'T'-key again.
- Othogonal - toggles between perspective and orthogonal modes of projection. In orthogonal mode objects appear at the same scale regardless of distance; in perspective mode further objects are scaled down.
- Show/Hide
- Box
- Molecule Info
- Atoms
- Bonds
- Symbols
- Hydrogen
- Carbon
- Mesh
- Features
|  |
| Supported File Formats |
|
The following file formats are supported by NCBI PubChem PC3D Viewer:
- PubChem Compound Files
-
PC3D Viewer supports generic NCBI PubChem molecule definition formats available for export at
//pubchem.ncbi.nlm.nih.gov:
- PubChem Compound
- provides one ore more conformer geometries for a single Compound
object from PubChem databases. PubChem Compound files are available at PubChem compound summary
pages for download. Note that some compounds do not have 3D geometries stored in PubChem databases. For such molecules
this viewer will not be able to render a 3D image. While loading these files the 3D Viewer performs
rotational and translational operations to the geometry of the molecule to align those with the intertial
frame of reference where Z-axis corresponds to the highest rotational momentum and X-axis corresponds
to the lowest. This aligns the molecules to appear flattest on the screen given the initial positioning of the viewport.
At the same time molecules are shifted to the their geometrial center (not the center of mass)
to create visually appealing, centered image of the chemical structure.
- PubChem Compounds
(plural) - provides a list of multiple 3D structures from different
Compound objects. Those objects are assumed to be aligned to each other by one of the conformer alignment
routines at PubChem. While loading such files the viewer will not attempt to cross re-align such molecules, but
will translate all those geomteries into an intertial coordinate system of the first compound in the list.
Thus, the cross-alignment present in the PubChem Compounds (plural) object is preserved.
- PubChem Substance
- provides one or more deposited structural records related to a single substance.
Usually these geometries are not computed by PubChem tools, but are provided by PubChem depositors as a part of their
initial deposition record.
Note that all three native formats can be packaged into files with extensions .pc3d, .asn or .asnt.
Native file types are registered by PC3D Viewer during installation on users PC as pc3d_compound. Corresponding
MIME type is registered as chemical/x-pc3d.
- SDF Files
- provides a single molecular geometry in a text ASCII format according to CTFile
format description. For comprehensive coverage on this topic see the provider's documentation.
SDF file may or may not contain bonding information in which case the bonding will be deduced from the coordinates of atoms and
covalent radia of correspodning elements.
- Compressed Molecular Files
- provides a compressed molecular file in gzip format.
- XYZ Model Files
- provides a single mlecular geometry in a text ASCII
formated file. Such file consists of a single header line with the number of atoms followed by an empty line, followed by
coordinate input lines. Each coordinate line has the chemical symbol followed by three coordinates (x,y,z).
Note, that XYZ files do not have bond information encoded in them, this is deduced from distances between atoms based on
the covalent radia of participating elements. This, although usually a good approximation, is not always accurate for chemicals
with non-covalent bonding.
| Keyboard/Mouse Operations |
|
- Rotation
X,Y rotations around the viewers x,y coordinate axes can be achieved by
simple mouse drags on a 3D viewer area. Moving the mouse vertically while left button is pressed will rotate the molecule around
user's equatorial x axis creating an impression of molecule following after the drag. Correspondingly, the horizontal mouse
movements will turn the molecule around user's axial y direction. Holding the Ctrl key while rotating the mouse pointer around the
center of the view will perform a rotation of a molecule around viewing z axis. Again, an attempt is made to create impression of
a molecule following mouse movements.
To achieve similar rotation results user may press Left and Right arrows keys on keyboard for equatorial rotations,
Up and Down for axial rotations and Home and End for rotations around the viewing direction. The rotations by keyboard
are performed by a larger increment of 15 degrees.
- Moving
dragging the mouse with left button and Shift key pressed will result in the molecule
being shifted from its centered location. The molecule will follow the mouse pointer in its direction. Note, that the center of molecular
rotations is always at the center of the view, not the center of the molecule. Therefore, after the molecule has beend shifted off-center,
further rotations will exaggerate the rotational displacement of the geometry.
- Scaling
rolling the mouse wheel backward/forward together with Ctrl key pressed
will result in moving the molecular geometry closer/further to the viewers camera. Thus, an impression of scaling up/down is achieved.
Note, that in one of the continous autoscale mode this operation has no effect since after each step the renderer will scale it back to the best fit.
The rotations, moves and scaling performed in one of the 3D viewers in tile mode will perform the operation not only in the viewer under the
mouse, but to the all viewers in all of the tiles. Thus, user can visually inspect all the molecules displayed in all of the smaller screens from
identical viewpoints. However, if this behavior is not wanted, the user may press and hold Alt key while performing the abovementioned
operations. Only the molecule under the mouse will then be affected by this operation.
- Aligment
to align the molecule to predefined positions along the main axes of coordinate
system user may press arrow keys while holding the Ctrl key. Ctrl+Left or Ctrl+Right will align along positive or negative
directions of user's equatorial axis; Ctrl+Up or Ctrl+Down aligns along axial direction; and Ctrl+Home or Ctrl+End align
along the viewing direction.
- Browsing
if multiple geometries are loaded into PC3D Viewer, pressing 'N' (next) or 'P' (previous)
will make the next/previous molecule visible and hide the currently visible conformer. This allows browsing the geometries of molecules one
by one sequentially.
User may choose to toggle on multiple conformers to see an overlap. In such case, next/previous operations
will hide only the very last conformer while showing the next in sequence. This adds a convenience of observing overlaps of
different conformers with reference geometries.
- Selection
clicking the left mouse button on any atom or its adjacent bonds will select that
atom. Information window will reflect by showing information about current selection. By selecting multiple atoms user may see not only the
coordinates of atoms, but the distances, flat angles and torsion angles between those. Note, that there can be
at most four atoms selected at the same time, selecting the fifth will deselect the first one. Clicking on
a selected atom again will deselect it.
|

